Document Actions
Aula 2 - Sequenciamento e Alinhamento genético
Nesta aula veremos um resumo sobre o sequenciamento e alinhamento genético e os principais softwares utilizados.
Parte 1 - Sequenciamento
Nesta primeira parte, veremos como é feito o sequenciamento genético.
O DNA e RNA são cadeias de polímeros compostas por uma pequena categoria de substâncias químicas similares. As unidades individuais são denomidadas nucleotídios ou bases. A utilização da base é diferente no DNA e no RNA. O código do DNA possui na sua composição os nucleotídeos A (adenina), T (timina), G (guanina) e C (citosina), enquanto que o RNA substitui o T pelo U (uracila). As bases se ligam aos pares sendo A-T e C-G.
Genômica - Ciência que estuda o genoma, ou o conjunto do material genético de um organismo.
Esse estudo é possível através do sequenciamento de DNA que determina a sequência nucleotídica. A partir daí, temos 3 tipos de projetos:
- DNA – seqüenciamento de estruturas do genoma ou de trechos destas. Ex.: Genoma humano
- Informações sobre regiões codantes (genes) e promotores.
- Mas gera sequências em regiões inter-gênicas (a princípio sem função)
- ESTs – sequenciamento de cDNA (DNA complementar), feitos à partir de bibliotecas de mRNA (RNA mensageiro) . Ex.: ESTs de cana-de-açúcar
- Informação direta sobre os genes e também sobre a expressão gênica.
- Mas genes pouco expressos são mais raros de serem sequenciados por essa técnica
- SAGE – sequenciamento de fragmentos em torno de 20 pb do cDNA.
SAGE fornece informação sobre a expressão gênica de forma mais eficiente que ESTs, mas é útil apenas quando o genoma completo do o organismo for conhecido
A situação ideal para um projeto genoma é sequenciar ambos DNA e cDNA
Veremos agora o método de sequenciamento Sanger um dos mais utilizados atualmente.
1) O DNA do núcleo de células é extraído por técnicas especiais. O DNA puro é então cortado em vários pedaços utilizando enzimas de restrição. Estas enzimas são muito específicas, isto é, cortam o DNA em locais conhecidos.
 |
| Quebra do DNA pela enzima em partes conhecidas |
2) Estes pedaços de DNA possuem as pontas conhecidas, pois foram cortados com enzimas conhecidas. As mesmas enzimas são utilizadas para cortar os plasmídios, que são DNA em forma de pequenos círculos. São fáceis de manipular porque, além de pequenos, possuem uma seqüência já determinada previamente. Assim, com o uso de outras enzimas, chamadas ligases, as pontas são unidas: ponta “A” liga-se com ponta “A” e ponta “B” liga-se com ponta “B”.
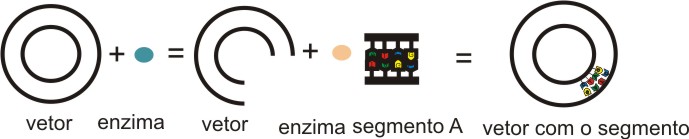 |
| Junção dos fragmentos de DNA ao vetor conhecido |
3) É desenhado um primer, um segmento de RNA com 1 a 60 nucleotídeos, uma sequência complementar do DNA. É através desse primer que uma enzima DNA polimerase, que sintetiza DNA apartir de um filamento complementar, formará as cadeias.
 |
| Desenho de um primer e a enzima DNA Polimerase |
4) São utilizados 4 bases dideoxynucleotídicas que além de possuirem uma propriedade fluorescente, em cores destintas, quando incorporadas, param a síntese do filamento complemetar.
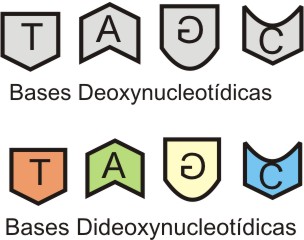 |
| Bases dideoxynucleotídicas |
5) Todos os elementos são aglutinados e a reação começa a formar cadeias de diversos tamanhos. Todas iniciando com o primer e terminados com uma base dideoxynucléica. Assim temos as cadeias que entrarão no sequenciador em uma solução de gel e a medida que as cadeias vão chegando ao final do gel são identificadas por um laser e codificadas num cromatograma.
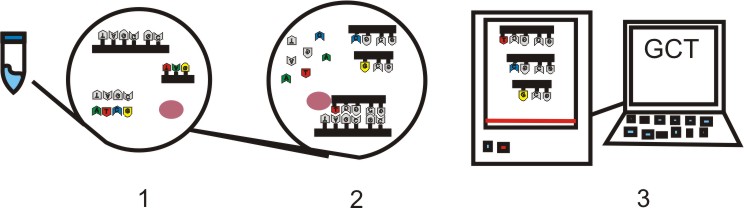 |
| Processo de formação das cadeias e entrada no sequenciador |
Animação explicando o sequenciamento Sanger.
Parte 2 - Alinhamento
Nesta parte veremos a étapa seguinte ao sequenciamento.
Após o sequenciamento, precisamos identificar e agregar informação a sequência. A melhor maneira de fazer isto, é comparar a sequência com sequências já conhecidas. Para isso, é feito o alinhamento par a par com um banco de dados.O que realmente importa é o grau real de similaridade ( estatisticamente significantes) temos:
- identidade = número que indica a quantidade de nucleotídeos alinhados
- similaridade = considera a probabilidade do alinhamento ter ocorrido por acaso (e-value). Considera todos os outros possíveis alinhamentos
- homologia = dividem a mesma ancestralidade com significado evolutivo
No contexto de evolução as sequências de DNA sofrem mutações. estas modificações locais entre os nucleotídeos podem ser :
- Inserções : inserção de uma base ou várias bases na sequência
- Deleções : deleção de uma base ou mais bases na sequência
- Substituições : substituição de uma base por outra
O conceito básico para a seleção de uma boa sequência de alinhamento é simples. As duas sequências são combinadas aleatoriamente. A qualidade da pontuação é avaliada e pontuada. Em seguida uma sequência é movida em relação a outra e a combinação é pontuada novamente, até que seja obtida a melhor pontuação de alinhamento.
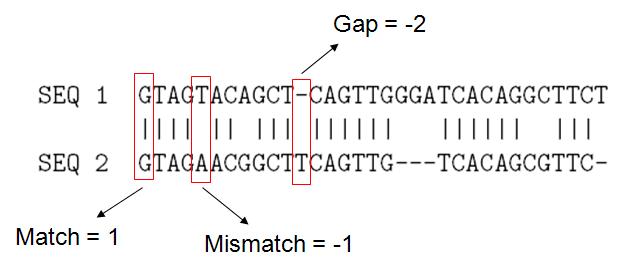 |
| Exemplo de alinhamento entre 2 sequências |
Gaps representam as inserções e deleções entre as sequências
O melhor alinhamento entre duas sequências é aquele que maximiza o score :
Score = #Matchs * (1) + #Mismatch * (-1) + #Gaps * (-2)
= 24 – 4 – 10 = 10
Temos os seguintes modelos de alinhamento:
- Alinhamento global - útil quando as duas sequências tem tamanhos próximos
- Alinhamento local - útil para alinhamento entre sequências de tamanhos diferentes e também para sequências com apenas alguns trechos conservados
- Alinhamento semi-global (ou pontas livres) - útil para encontrar sobreposições de fragmentos de sequenciamento
Parte 3 - Softwares
Veremos agora os softwares responsáveis por cada etapa da análise.
O arquivo de saída do sequenciador é um cromatograma que contém a sequência e a qualidade das bases.
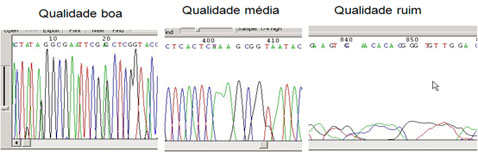 |
| Exemplo de um cromatograma e a qualidade das bases |
- o Phred será o primeiro software que iremos utilizar. Ele analisa o cromatograma e dá uma nota para cada base baseado em sua qualidade.
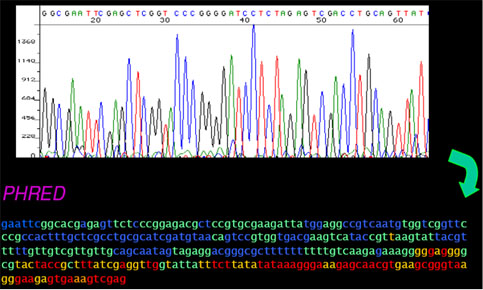 |
| Cromatograma e a saída do phred |
O phred utiliza esta formula para atribuir a nota:
q = -10 log10 (P)
onde q é a qualidade e P a probabilidade de encontrar uma base errada. Uma nota phred 20 implica em uma base errada em cada 100, e uma nota 30 uma base errada em cada 1000.
- Após obter o arquivo com as notas de cada base, devemos identificar o vetor que foi usado no sequenciameno. Para isso usamos o Cross_match. Ele faz uma comparação entre as sequências e mascara a região do vetor na sequência. Isto é, substitui os nucleotídeos vindos do vetor pela letra X,
| >Seq1 XXXXATGCGCATAGCATAGGGACATCATACATTTTACACACAAGAGACAGACGAT ACTACATGTCATGACTACXXXXXXXXXXXXXXX |
| Exemplo de saída do cross_match |
- Com o vetor identificado, vamos realizar o alinhamento entre as sequências. Para essa função usaremos o Cap3. Ele realiza alinhamentos semi-global e gera arquivos contendo os singlets e os contigs.
- Agora que ja temos os alinhamentos entre as sequências, faremos outro alinhamento agora contra um banco de dados. Para isso usamos o Blast. Ele utiliza alinhamento local para confrontar as sequências com um banco de dados conhecido. Assim temos como saída, com que tipos de estruturas conhecidas a sequência é mais similar.
| BLASTX 2.2.17 [Aug-26-2007] Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs", Nucleic Acids Res. 25:3389-3402. Query= Contig1 (896 letters) Database: All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF excluding environmental samples from WGS projects 6,307,426 sequences; 2,155,784,383 total letters Searching..................................................done Score E Sequences producing significant alignments: (bits) Value ref|XP_312613.4| AGAP002350-PA [Anopheles gambiae str. PEST] >gi... 201 7e-50 ref|XP_001688392.1| AGAP002350-PB [Anopheles gambiae str. PEST] ... 201 7e-50 >ref|XP_312613.4| AGAP002350-PA [Anopheles gambiae str. PEST] gb|EAA44956.4| AGAP002350-PA [Anopheles gambiae str. PEST] Length = 383 Score = 201 bits (510), Expect = 7e-50 Identities = 95/111 (85%), Positives = 103/111 (92%) Frame = -1 Query: 857 VKLETEKYDLEERQKRQDFDLKELKERQKQQLRHKAFKKGVDPEALTGKYPPKIQVASKY 678 VKLETEKYDLEERQKRQD+DLKELKERQKQQLRHKA KKG+DPEALTGKYPPKIQVASKY Sbjct: 206 VKLETEKYDLEERQKRQDYDLKELKERQKQQLRHKALKKGLDPEALTGKYPPKIQVASKY 265 Query: 677 EGRVDTRSFDDKKKIFEGGFETLTKEFFEKVWHEKKEEFGGCQKTKLPKWF 525 E RVDTRS+DDKKK+FEGGF+TL KE EK W E+KE+FGG QK+KLPKWF Sbjct: 266 ERRVDTRSYDDKKKLFEGGFDTLNKEVLEKQWAERKEQFGGRQKSKLPKWF 316 |
| Exemplo de um arquivo de saída do Blast |
Referências
Falsarella, Marcelo - Slides do curso de Bioinformática aplicada a genéticaGibas, Cynthia – Desenvolvendo Bioinformática